I was always interested in the applied bioinformatics. Therefore, some of my PhD projects focused on the Human Gut Microbiome, which is an essential factor in antibiotic resistance emergence in the context of medicine. The gut is full of nutrients and packed with bacteria, the gut constitutes a perfect environment for speeding up bacterial evolution. Each antibiotic therapy drives the emergence and retention of antibiotic resistance genes within the human gut microbiome. The group of Prof. Daniel Huson at the University of Tuebingen specialised in generating algorithmic tools to analyse ever growing in size and complexity metagenomics datasets.
The human gut microbiome has become an important contributor to human health. Limited access to sequencing and clinical data makes data-driven meta-analyses challenging. Thus, no comprehensive resource exists to interpret gut microbiome composition.
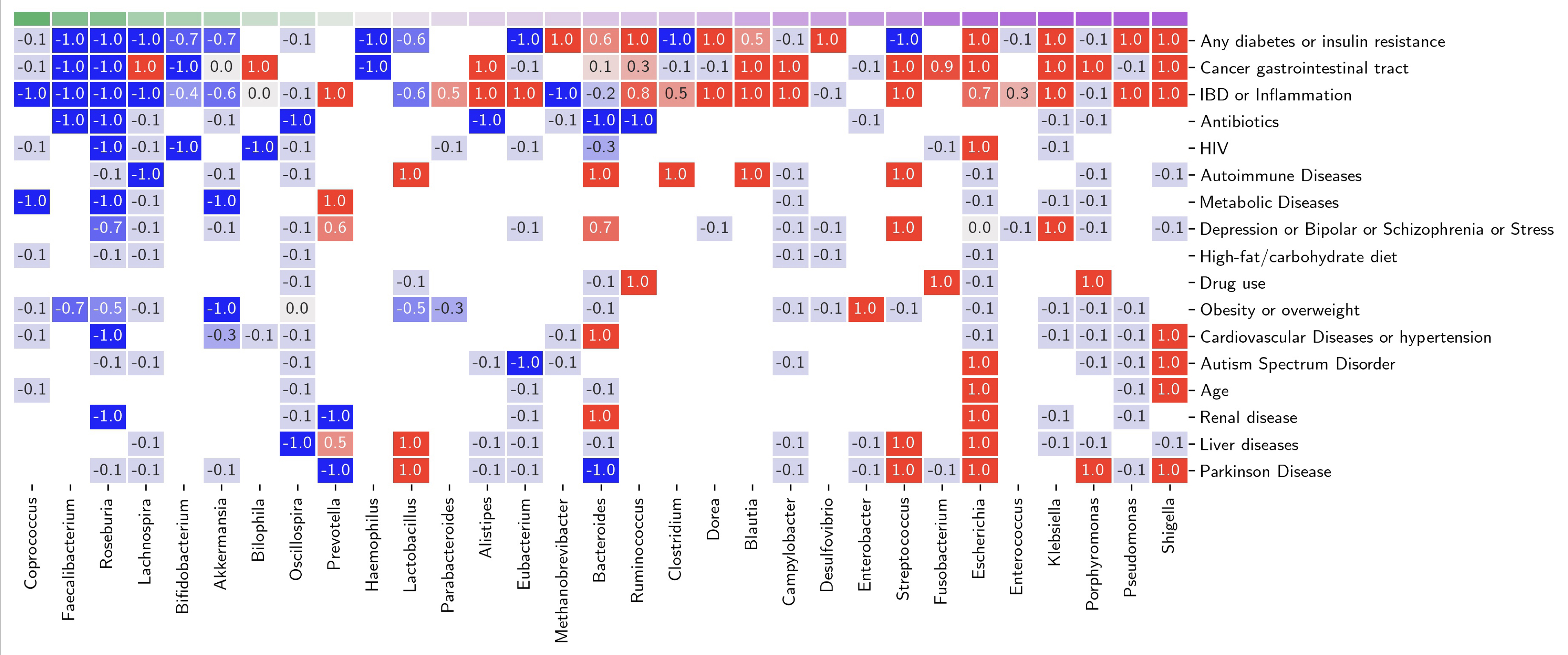
We conducted a streamlined meta-analysis of abstracts from observational studies comparing patients to healthy controls, focusing on the relationships between gut microbiome genera and various factors. Extracted data included genus, disease, association (positive/negative encoded as +1/-1), methods, and population size. Results were summarized by averaging relationship values for each genus-condition pair. Each genus’s inflammation score was calculated as the sum of its average scores and applied to interpret the gut microbiome composition of six healthcare workers before, during, and after COVID-19, using 16S rRNA sequencing. For each sample, the Shannon diversity index and an inflammation ratio (the abundance of pro-inflammatory versus anti-inflammatory genera, based on the inflammation score) were computed.
A total of 4,015 abstracts were screened, including 276 publications and 1,162 data points across 106 genera (68 described in more than two studies) and 91 diseases. Despite its simplicity, this analysis reveals clear patterns linking specific genera to diseases and other factors. For example, Faecalibacterium, Roseburia, and Bifidobacterium are associated with low inflammation scores (Figure 1), while Shigella, Pseudomonas, and Klebsiella are linked to high inflammation scores. Figure 2 demonstrates that the inflammation ratio better captures the nature of gut microbiome changes than the widely used Shannon diversity index.
Together with prof. Elda Righi, we investigated impact of the COVID-19 on the gut microbiome. Even a mild infection with a SARS-CoV-2 disturbs the gut microbiome deeply and rapidly. That, together with hospitalisation constitutes another possible long term negative effect of COVID.
In this project, we studied only two participants for the six timepoints: before ciprofloxacin therapy, in the first, third, 6th day of the treatment - then 2 days after the therapy ended and 28 days after. The gut microbiome samples were sequenced in two different modes: ultra-deep whole-genome shotgun metagenomics on the MiSeq. A phage-only sequencing used a specialised protocol to extract the viral-like particles first and in the second step the DNA. This setup enabled to study how the structure of the gut microbiome reconstructs after the powerful disturbant that is the therapy with the fluoroquinolone.
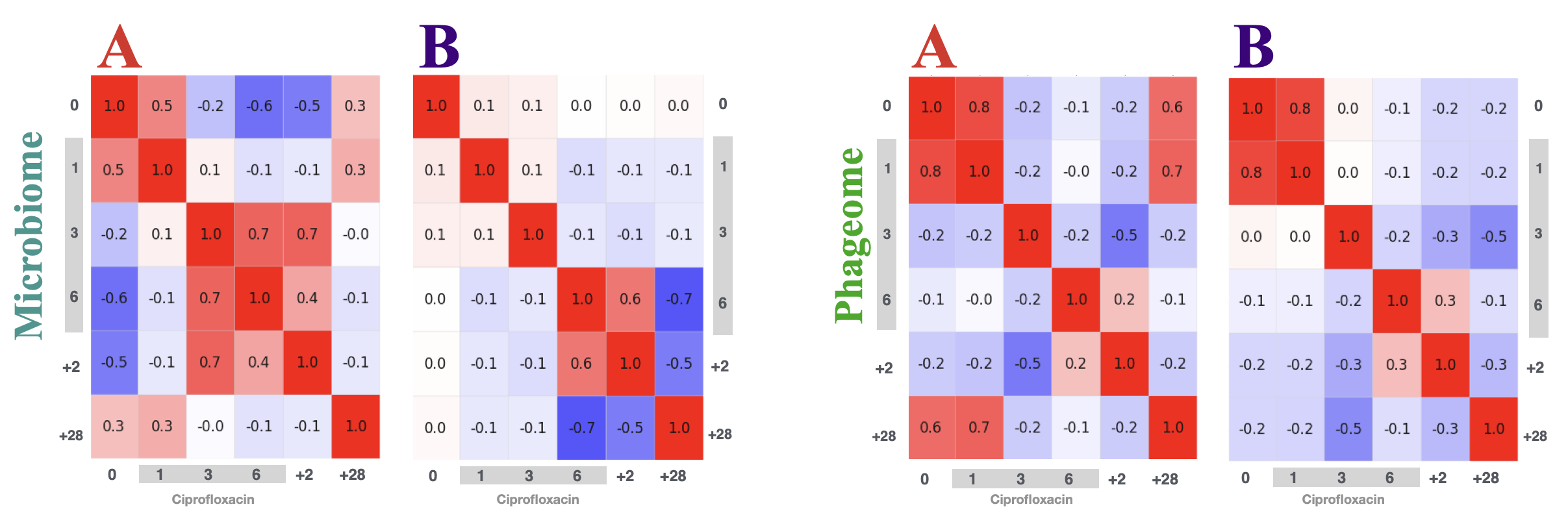
The depth of sequencing in the WGS allowed me to analyse through the assembly - I've assembled all of the samples and then re-mapped the reads to the fragments. The fragments were annotated with an array of the tools. Thus, each feature from the annotation tools could be tied to the overall prevalence within the sample by transforming and summarising the number of reads that mapped to the fragnemnt.
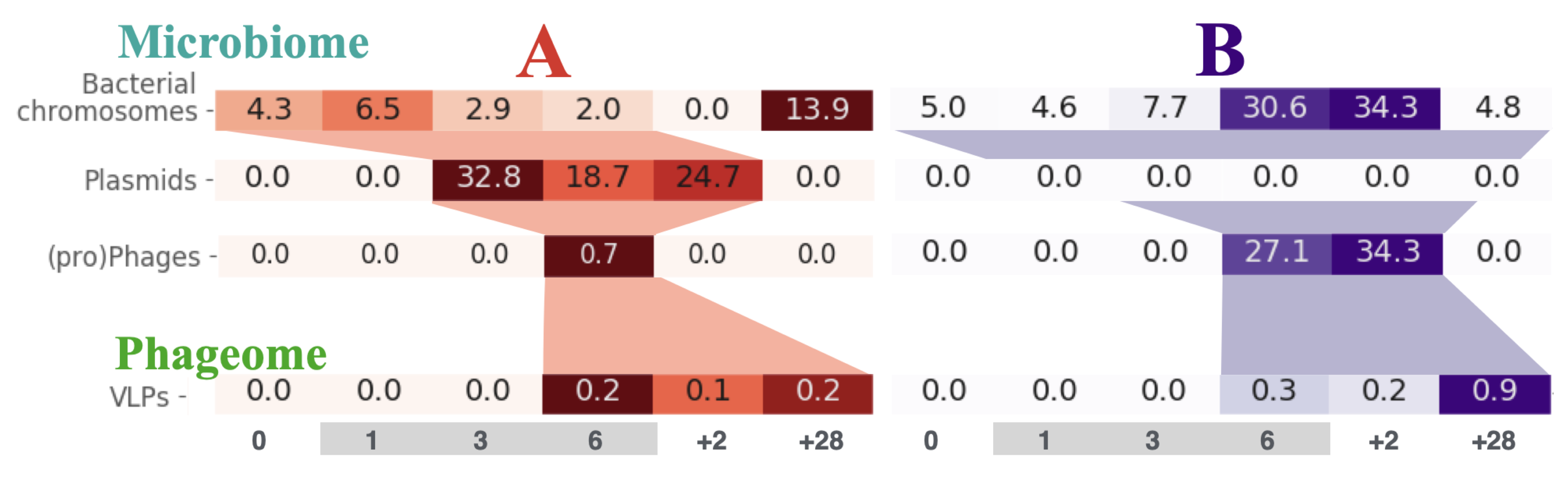
Although, the details on how the resistance spreads between bacteria within the human gut remain unknown, as does the role of horizontal gene transfer in this process. We presented a novel approach to the analysis of time-series whole-genome metagenomic sequencing data. This involves partitioning the scaffolds from the metagenomic assembly into groups corresponding to bacterial chromosomes, plasmids and those with prophages and transposons. Using specialized sequencing of the bacteriophages we were able to track the flow of ciprofloxacin resistance genes from bacterial chromosomes, through the plasmids, to prophages and phages (Figure).
Microbiome analysis and especially metagenomics was one of the leading research fields of the prof. Huson's group where I did my PhD. We developed tools (MEGAN) and programmatic challenges of the microbiome analysis. My main contribution was setting up a MEGAN user community, and providing the basic tutorials and content related the the common questions.
The Whole-Genome metagenomics provides information about the relative abundance of the taxa and the proteins of specific function across an array of functional classifications. However, short reads do not provide information on what functions and taxa are together. Long read technologies will bring breakthroughs in metagenomics analysis. However, the long read flipped the algorithmic paradigm of the metagenomic analysis - as now the reads now contain multiple hits from the database, and that the NanoPore is characterized with a different errors that can break the aligners. In the Huson lab we worked on bringing the long reads into metagenomic sequencing and MEGAN. All of the tools needed updating to the new method. From both short Illumina sequencing and NanoPore sequencing of the content of the bioreactor, we were able to extract several full circular genomes of the bacteria, and annotate them with an array of the genes. My role in this publication was analysing and plotting the assembled and corrected genmoes: